發達國家向非發達國家發射的“三枚煙霧彈”
日期:2015/9/2
文/謝沐風(上海市食品藥品檢驗所)
自2013年11月在《中國醫藥工業雜志》發表“仿制藥研發中有關物質研究思路之我見”后,收到大量同仁來電來函交流。積累一年多心得,撰寫此續篇,希冀能將某些觀點闡述得更為清楚明了,進而為我國仿制藥研發中雜質研究思路與控制策略提供一種更為客觀理性的認知,為科學厘清雜質對于藥物臨床使用的價值與意義呈上綿薄之力,為正確認清本行業高科技的核心與實質獻計獻策,為將我們的有限資源用到研發關鍵之處指明方向。
1. 從宏觀上解讀雜質
1.1. 雜質與藥物不良反應的關系
……
1.2. 雜質與藥物質量控制的關系
……
1.3. 發達國家向非發達國家施放的“煙霧彈”
國內對雜質研究之所以“著力猛攻、一網打盡”,還與盲目迷信國外文獻資料有關。由于制藥行業的利潤實在太過豐厚,所以吾等千萬不要幼稚地以為“老外們會很傻/很天真地和盤托出”,相反原研企業會千方百計地在仿制藥研發與申報的征途中設置一些障礙或施放一些“煙霧彈”來迷惑我們……。舉例如下:
1.3.1. 第一枚煙霧彈 —— 雜質
秉承制劑質量標準僅關注降解雜質的原則,在前些年的國外藥典和進口質量標準中很多制劑品種無有關物質檢查項,這是科學合理的:經研究當“原料藥制成0天制劑”和“0天制劑在加速試驗6個月/長期試驗6個月/貨架期內”,這兩個環節中雜質均無變化(不是沒有、是無變化),便可在制劑質量標準中無需制訂有關物質檢查項,從而實現發達國家早在多年前就開始提倡和踐行的“綠色檢驗”理念——為減少環境污染與試劑排放,檢驗工作應盡可能做到事半功倍、一針見血。
但近些年,這些標準中卻開始制訂并收載大量雜質,且有愈演愈烈之態勢,乍看起來甚是“高標準。嚴要求”,但本人認為這是在施放“煙霧彈”、目的是引仿制者誤入歧途。解讀如下案例:
某一普通口服片劑進口質量標準(規格為1.0mg~0.125mg)中收載了12個已知雜質,并采用了極為復雜的色譜系統進行測定,還引入校正因子逐一準確計算,并制定了每一雜質限度。秉承上述原則,該藥物是不可能在效期內有12個降解雜質產生的。果然,經對數批貨架期內原研制劑樣品予以檢測或取1批樣品進行加速試驗6個月測試,絕大部分都是未檢出(報告限以下)、僅有1~2個雜質在報告限以上、鑒定限以下,根本無需研究。可令人遺憾的是:國內眾多同仁將進口質量標準/國外藥典奉為“圭臬”、當作“圣旨”,花費大量金錢與資源去設法獲得那12個雜質,最終經研究,在仿制原料藥和仿制制劑中均是未檢出,因我國合成能力極強,完全有能力將原料藥中雜質全部去除;再加上主成分的自身穩定性很好、制成制劑后雜質也不會有變化。
針對該品種,本人推薦研發思路如下:直接采用既有質量標準中的色譜條件、無需驗證,第一步/測定三批原研制劑樣品,并經對雜質譜剖析、無含量不斷增加雜質;第二步/測定三批規模化生產的仿制原料藥與仿制制劑樣品,無制劑鑒定限以上雜質,且無含量不斷增加雜質;第三步/根據以上研究結果,最終在制劑的質量標準中無需制訂有關物質檢查項,原料藥質量標準也僅是粗略地制訂單雜和總雜即可,根本無需制訂那么多雜質。甚至采取更為大膽的作法——由于主成分規格很小,且口服,每日最多3片,故雜質與殘留溶劑均無需研究,只需在申報材料中闡明:經推算這些物質的每日最高攝入量均小于每日臨床安全攝入限度即可,從而做到專業的最高境界——有所為有所不為。
還有一進口外用涂抹皮膚的乳膏劑質量標準,主成分規格依然很小、卻制訂了8個已知雜質。本人認為這些均是原研企業對仿制者施放的煙霧彈,目的就是阻礙仿制進程、迷惑研發方向的表現。
至于如何看待國外藥典原料藥項下羅列的眾多雜質,且毫無保留地給出了結構式、花錢還可購買來(價格甚是不菲),那更是仁者見仁、智者見智。但此處筆者想提醒的是:目前全世界絕大多數原料藥均已由吾等發展中國家生產后出口至ICH國家(把霧霾和污染留給了我們自己),發達國家要做的:是將這些“已把雜質幾乎摳沒”的原料藥,經過特殊的、保密處理后(例如重結晶后制成目標晶型、粒度分布、顆粒形狀、比表面能、晶格能、固有溶出速率等特性)制成制劑、再銷往全世界獲取高額利潤,當然質量絕對毋庸置疑。
1.3.2. 第二枚煙霧彈 —— 溶出度試驗
各劑型均有關鍵性評價指標,在ICH Q6文件(規范:新原料藥和新藥制劑的測試方法和認可標準:化學物質)和世界衛生組織藥品標準專家委員會第43次技術報告一書中均有相關闡述。
發達國家針對這些關鍵性評價指標,要么隱含(僅在嚴格保密的、企業內控質量標準中才有),要么即便公開、也大多為寬松的試驗參數與限度范圍。如對于口服固體制劑,最為核心的是溶出度試驗。本人曾接觸過上百個原研制劑多條溶出曲線剖析測定結果,發現相當一部分國外藥典和進口質量標準中的試驗參數都極為寬松、不具區分力(如采用高轉速、高濃度表面活性劑、或是高溶解度的溶出介質)。蓋因該試驗太重要,連發達國家在制訂ICH Q系列質量管理文件時都未有專門的指導原則進行闡述,唯《日本溶出曲線數據庫》收載的實驗條件是真實的、最具區分力的條件。
這里需闡明:日本自上世紀九十年代起就十分重視體外溶出度試驗,因日方很早就意識到生物等效性(BE)試驗由于僅采用年輕男性作為受試者有其局限性(故BE試驗不是“金標準”),在無法采用各種人群大標本試驗的前提下,只能通過對體外溶出度試驗的嚴格要求來促使仿制制劑內在品質“無限趨近”原研制劑。為此,日本在1997年出版的《仿制藥生物等效性試驗指導原則》中就已有了詳盡的溶出度試驗研究要求,并占據了一半篇幅。同時,迄今為止日本官方已建立起700多個品種的原研制劑四條溶出曲線數據庫,并予以了全面的真實公開,且該項工作還在不斷進行中……。
而歐美的公開資料中則大多宣稱:體內生物利用度試驗才具有最終決定性,不太強調體外溶出度試驗。但通過解讀他們公開文獻中的溶出度試驗參數,發現多數不具區分力、甚至干脆不公開(如英國藥典收載的制劑很少,甚至有些難溶性口服固體制劑竟然沒有溶出度試驗,緩控釋制劑更是一個品種也無釋放度試驗)的作法說明:這是一種韜光養晦、瞞天過海的表現。如我們按此種不具區分力的實驗條件去研制仿制制劑,則多半會出現“部分仿制藥(主要是那些有制劑難度的品種:難溶性口服固體制劑、腸溶制劑、緩控釋制劑、pH值依賴型制劑、治療窗狹窄藥物制劑等)對于部分患者(尤中老年人)安全無效、吃了白吃”的結果。不深諳溶出度評價,就不知努力的方向與差距所在,因溶出度試驗才是撬動制劑內在品質不斷提升的“那根杠桿”,只有掌握科學客觀的評價法,才能做出與原研制劑內在品質無限趨近的高品質仿制藥來,這將充分體現“標準就是生產力”的精神。
1.3.3. 第三枚煙霧彈 —— 潛在基因毒性雜質
確實發生過類似事件,但絕對是“極小概率”。我國擷取后營造出的學術氛圍就猶如發現了一大“寶藏”,提高至“報告限以上雜質就應考慮,甚至潛在雜質也要考慮”;而“一旦潛在、就無窮無盡了”。近三年,針對該類雜質開展了轟轟烈烈的研究,花費巨資購買了大量進口高精尖檢測設備,而最終樣品檢測結果很多都是“未檢出”。
其實,只要觀測仿制原料藥的“起始物、副產物、試劑、配位體、催化劑”等這些物質是否具備文獻報道的“基因毒性雜質官能團結構式”,并針對規模化生產的仿制原料藥三批樣品中檢出的鑒定限(0.10%)以上特有雜質(即原研制劑中不存在的)是否具備這些結構式,如具備再進行相應研究便是,切忌“大驚小怪”、猶如“驚弓之鳥”。
現今,隨著科學發展人們認知到:連藥物自身都會給人體帶來致癌毒性。所以,強烈建議我國藥品審評中心(CDE)的審判員和藥物研發者均無需過于謹小慎微、患得患失。
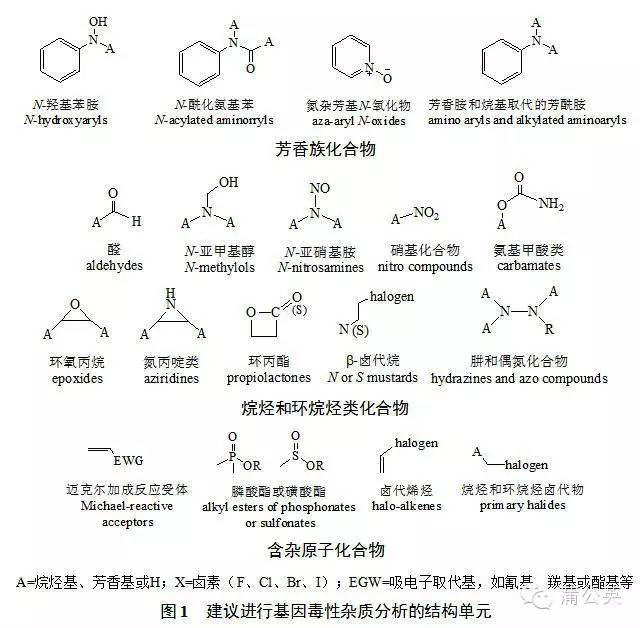
A為烷烴基、芳香基或H;X為鹵素,包括F、Cl、Br、I;EGW為吸電子取代基,如氰基、羰基或酯基等
圖1 具備以上結構單元的雜質、建議進行基因毒性雜質的分析和說明
1.3.4. “其他煙霧彈”
今后一定還會有其他“煙霧彈”發射過來,如近期興起的“金屬雜質研究”等。
1.3.5 無獨有偶
1.3.6. 應對策略
首先我們應理解這種行為無可厚非,這是由本行業的特殊性所決定的(“N高” —— 高投入、高利潤、高附加值、高端制造業)。我們作為發展中國家,在仿制過程中,要具備“公開的就不是高科技、看不到的才是高科技”之辯證唯物觀點。具體建議如下:
(1) 針對進口質量標準 相關藥檢機構在進行復核時,應要求外企提供詳盡的檢測法驗證資料,即應著重審閱質量標準中的各試驗參數是如何建立的、關注其邏輯推導過程,是經怎樣的研究內容與試驗結果確定的,這才是審核重點與核心;如未盡詳細,應要求對方持續提供。如溶出度試驗,為何申報中制訂了100rpm高轉速和該溶出介質?是否研究過50和75轉溶出行為以及其他介質的溶出情況后綜合考慮確定?是否有故意放寬、讓自我產品永遠合格的嫌疑。又比如有關物質,為何訂入這些多個雜質和這么低的限度,制訂的依據是否充分等等。
(2) 針對公開的各國藥典與文獻資料 切忌照搬照抄、盲目迷信。CDE早在2003年就已提出“仿產品不是仿標準”的指導思想,就是希冀眾人在具體研制時能獨立思考、冷靜分析。遺憾的是;很多同仁認為該宗旨中的“標準”僅是指國內質量標準、不包括國外標準/文獻,導致被這些標準迷惑、誤入歧途。本人從事藥物分析工作17年,深感:發達國家的分析檢測水平很一般、有時甚至拙劣,他們強在藥劑、尤工業藥劑學上。
(3) 總之,無論如何都應獲取至少三批原研制劑,深度剖析其關鍵質量特性參數后再來指引自我仿制藥的開發和制訂質量標準等事宜,這才是研發的根本出發點,也唯有如此才能做到“知己知彼、百戰不殆”。
信息來源:蒲公英
|


