CFDA發布《化學藥品注冊分類改革工作方案》,分類作了顛覆性調整!
日期:2016/3/6
3月4日,國家食品藥品監督管理總局發布《關于發布化學藥品注冊分類改革工作方案的公告(2016年第51號)》,并自公告發布之日起實施。
可以看出,相較于現行的《藥品注冊管理辦法》,第51號公告中對于化學藥的分類作了顛覆性的調整,與此前的公開征求意見稿中的分類也多有不同。
《公告》對化學藥品注冊分類類別進行調整,化學藥品新注冊分類共分為5個類別,具體如下:
1類:境內外均未上市的創新藥。指含有新的結構明確的、具有藥理作用的化合物,且具有臨床價值的藥品。
2類:境內外均未上市的改良型新藥。指在已知活性成份的基礎上,對其結構、劑型、處方工藝、給藥途徑、適應癥等進行優化,且具有明顯臨床優勢的藥品。
3類:境內申請人仿制境外上市但境內未上市原研藥品的藥品。該類藥品應與原研藥品的質量和療效一致。
原研藥品指境內外首個獲準上市,且具有完整和充分的安全性、有效性數據作為上市依據的藥品。
4類:境內申請人仿制已在境內上市原研藥品的藥品。該類藥品應與原研藥品的質量和療效一致。
5類:境外上市的藥品申請在境內上市。

《藥品注冊管理辦法》(2007版):藥品注冊申請包括新藥申請、仿制藥申請、進口藥品申請及其補充申請和再注冊申請。新藥申請,是指未曾在中國境內上市銷售的藥品的注冊申請。對已上市藥品改變劑型、改變給藥途徑、增加新適應癥的藥品注冊按照新藥申請的程序申報。仿制藥申請,是指生產國家食品藥品監督管理局已批準上市的已有國家標準的藥品的注冊申請。



■李瑤 整理
■編輯 陳雪薇
總局關于發布化學藥品注冊分類改革工作方案的公告(2016年第51號)
根據2015年11月4日第十二屆全國人民代表大會常務委員會第十七次會議審議通過的《關于授權國務院在部分地方開展藥品上市許可持有人制度試點和有關問題的決定》,國家食品藥品監督管理總局制定了化學藥品注冊分類工作改革方案,已經國務院同意,現予以公告,并自公告發布之日起實施。
化學藥品注冊分類改革工作方案
為鼓勵新藥創制,嚴格審評審批,提高藥品質量,促進產業升級,對當前化學藥品注冊分類進行改革,特制定本工作方案。
一、調整化學藥品注冊分類類別
對化學藥品注冊分類類別進行調整,化學藥品新注冊分類共分為5個類別,具體如下:
1類:境內外均未上市的創新藥。指含有新的結構明確的、具有藥理作用的化合物,且具有臨床價值的藥品。
2類:境內外均未上市的改良型新藥。指在已知活性成份的基礎上,對其結構、劑型、處方工藝、給藥途徑、適應癥等進行優化,且具有明顯臨床優勢的藥品。
3類:境內申請人仿制境外上市但境內未上市原研藥品的藥品。該類藥品應與原研藥品的質量和療效一致。
原研藥品指境內外首個獲準上市,且具有完整和充分的安全性、有效性數據作為上市依據的藥品。
4類:境內申請人仿制已在境內上市原研藥品的藥品。該類藥品應與原研藥品的質量和療效一致。
5類:境外上市的藥品申請在境內上市。

二、相關注冊管理要求
(一)對新藥的審評審批,在物質基礎原創性和新穎性基礎上,強調臨床價值的要求,其中改良型新藥要求比改良前具有明顯的臨床優勢。對仿制藥的審評審批,強調與原研藥品質量和療效的一致。
(二)新注冊分類1、2類別藥品,按照《藥品注冊管理辦法》中新藥的程序申報;新注冊分類3、4類別藥品,按照《藥品注冊管理辦法》中仿制藥的程序申報;新注冊分類5類別藥品,按照《藥品注冊管理辦法》中進口藥品的程序申報。
新注冊分類2類別的藥品,同時符合多個情形要求的,須在申請表中一并予以列明。
(三)根據《中華人民共和國藥品管理法實施條例》的有關要求,對新藥設立3—5年監測期,具體如下:
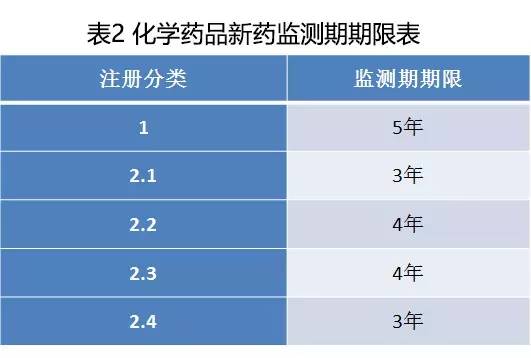
(四)本方案發布實施前已受理的化學藥品注冊申請,可以繼續按照原規定進行審評審批,也可以申請按照新注冊分類進行審評審批。如申請按照新注冊分類進行審評審批,補交相關費用后,不再補交技術資料,國家食品藥品監督管理總局藥品審評中心要設立綠色通道,加快審評審批。符合要求的,批準上市;不符合要求的,不再要求補充資料,直接不予批準。
(五)新注冊分類的注冊申請所核發的藥品批準文號(進口藥品注冊證/醫藥產品注冊證)效力與原注冊分類的注冊申請核發的藥品批準文號(進口藥品注冊證/醫藥產品注冊證)效力等同。
(六)國家食品藥品監督管理總局組織相關部門細化工作要求,做好受理、核查檢查、技術審評及制定、修訂相關國家藥品標準等工作。
(七)《藥品注冊管理辦法》與本方案不一致的,按照本方案要求執行。
信息來源:醫藥經濟報
|


